Linearized Pair-Density Functional Theory
Generally speaking, we are interested in understanding how matter and light interact with each other because of its applications to UV-damage (and sunscreens), photosynthesis, and catalysis. There is a lot of work focused on developing electronic structure methods which can accurately compute the potential energy surface of systems, both the ground and excited state. One such method, which is particularly attractive, is multiconfiguration pair-density functional theory (MC-PDFT) due to its speed and accuracy. MC-PDFT is similar to Kohn-Sham DFT in that it computes the energy for a particular state using a energy expression which is expressed as a functional of the density.
\begin{equation} E^\mathrm{PDFT} = h^q_p \gamma^p_q+ \frac{1}{2}g_{pr}^{qs}\gamma^p_q\gamma^r_s + E^\mathrm{ot}[\rho, \Pi] + V_\mathrm{nuc} \end{equation}
While this is good, it unfortunately does not adequately model the electronic structure/potential surfaces where states of the same symmetry get close together in what are called conical intersections or near-avoided crossings. These regions of the potential surface are very common when modeling photochemistry, and are the key drivers for internal conversion processes. As such, I have worked on developing a multi-state PDFT (MS-PDFT) called linearized PDFT (L-PDFT). L-PDFT works by functionalizing an effective Hamiltonian on a particular model-space (or set of densities). Essentially, the L-PDFT Hamiltonian comes from Taylor expanding the MC-PDFT energy expression to first order around the state-average densities, and extracting the effective linear operator.
\begin{equation} H^\mathrm{L-PDFT} = \left(h^q_p + V^q_p + \mathcal{J}[\check{\gamma}]^q_p\right)E^p_q + \frac{1}{2}v_{pr}^{qs}e_{qs}^{pr} + h^\mathrm{const} \end{equation}
Diagonalizing this operator with in an appropriate model space yields the correct potential energy surface topology near conical intersections and near-avoided crossings. Additionally, it is formally faster than MC-PDFT since it only has a single quadrature calculation. Furthermore, it is as accurate as MC-PDFT for vertical excitations whereas other MS-PDFT methods are not necessarily.
L-PDFT also has the advantage of having a bon afide operator making it more suitable for future development. You can check out the first paper describing the method here.
Most of my Ph.D. work has been on developing out L-PDFT for applications in various areas of photocatalysis and photochemistry. My development (at least initially) is within the PySCF-forge python extension package for PySCF and SHARC, though I hope to integrate it into some other quantum chemistry software packages.
L-PDFT Properties
The following is a list of properties that are currently developed for L-PDFT (as of Aug 2025).
- L-PDFT total electronic energy calculations for wave functions of various
types [1].
- State-averaged complete active space configuration interaction (SA-CASCI)
- State-averaged complete active space self-consistent field (SA-CASSCF)
- SA-CASCI/SA-CASSCF with “mixed” solvers of different spins and/or point group symmetries [2]
- L-PDFT can be used with various translated or fully-translated Kohn-Sham density functional theory (KS-DFT) functionals as defined in libxc [1].
- Additional Wave Function Properties
- Analytic nuclear gradients (with non-hybrid functionals only) for:
- SA-CASSCF wave functions [3]
- Analytic nuclear gradients (with non-hybrid functionals only) for:
Vertical Excitations Between Symmetry Irreps
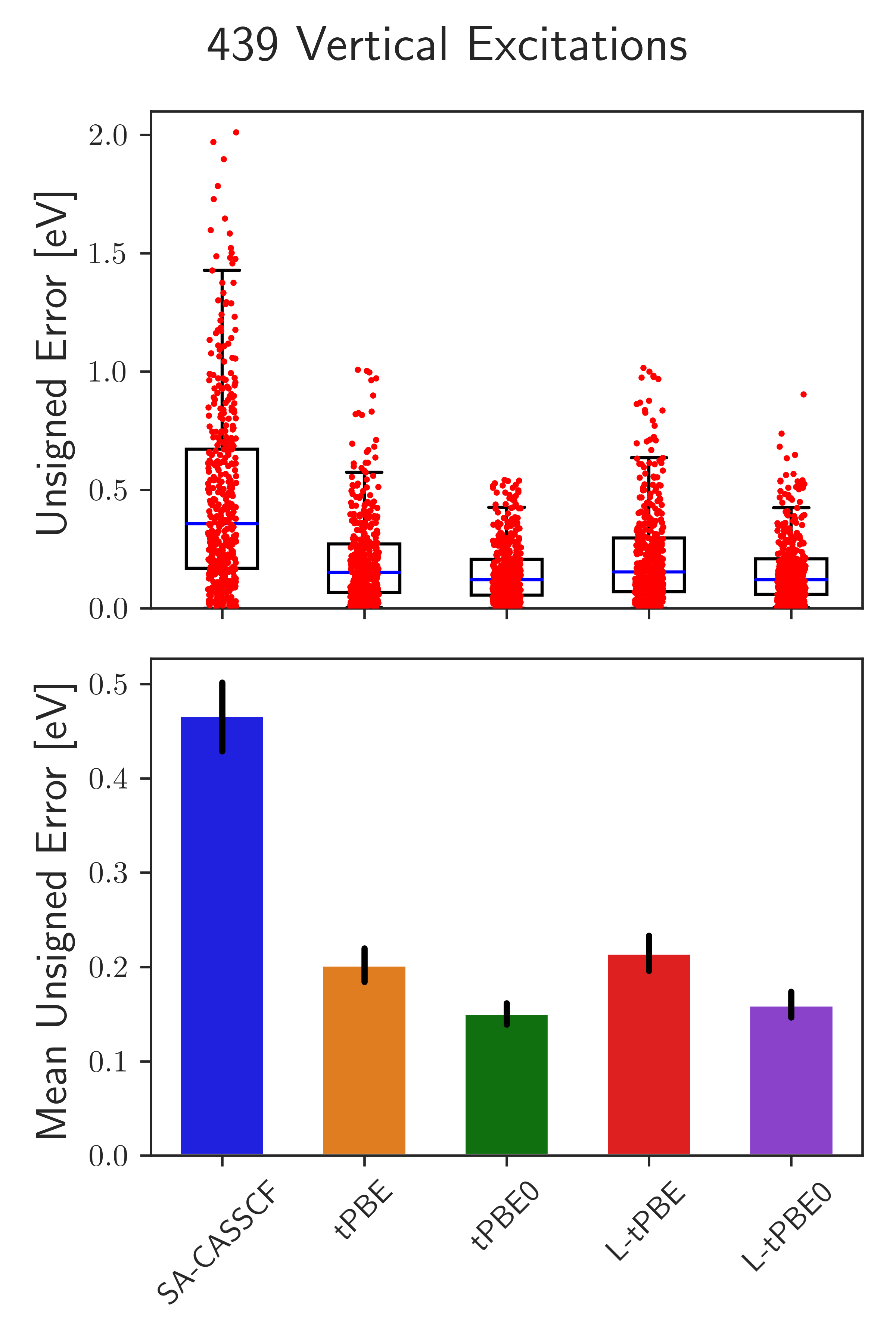
We have subsequently defined how to perform vertical excitations between states of different spin or spatial symmetry within L-PDFT. Here, we expand around the state-average densities across all of the symmetry irreps. In this way, we showed that L-PDFT performs similarly to MC-PDFT on the QUESTDB database illustrating how L-PDFT can be used in regions of strong and weak state-interaction [2]. Coupling this with L-PDFT being faster than MC-PDFT, L-PDFT seems to be a promising method for photodynamics in the future.
L-PDFT For Photodynamics
Given the recent implementation of analytical nuclear gradients for L-PDFT based on a SA-CASSCF wave function, we performed nonadiabatic molecular dynamic simulations of cis-to-trans azomethane [4]. Our results found that L-PDFT dynamics were stable with no trajectories crashing, though a few trajectories did not conserve total energy (due to the active space problem). In general, our results were in agreement with much more expensive multi-state complete active space second order perturbation theory (MS-CASPT2) while only being a little bit more expensive that SA-CASSCF. In general, this was the first time a pair-density functional method was used to perform molecular dynamics for an internal conversion process, and illustrates the utility of L-PDFT to study a wide variety of photochemical and photo-induced processes.
Acknowledgements
This work was performed under the guidance of Professor Laura Gagliardi during my time as a Ph.D. student at UChicago. Additionally, this work was supported in part by the National Science Foundation Graduate Research Fellowship. I also acknowledge the University of Chicago’s Research Computing Center for their support of this work. This work was also partially supported by the University of Chicago’s Department of Chemistry though the McCormick Fellowship (Sept 2021) and Olshansky Graduent Student Travel Award (June 2023), as well as the University of Chicago Physical Science’s Division Eckhardt Graduate Fellowship (2021).
Code Developed for this Project
- Linearized Pair-Density Functional Theory in PySCF-forge
- Born-Oppenheimer Molecular Dynamics Package in PySCF
- SHARC-PySCF interface for nonadiabatic molecular dynamics using L-PDFT
References:
M. R. Hennefarth, M. R. Hermes, D. G. Truhlar, and L. Gagliardi, “Linearized pair-density functional theory,” J. Chem. Theory Comput. 19, 3172-3183 (2023).
M. R. Hennefarth, D. S. King, and L. Gagliardi, “Linearized pair-density functional theory for vertical excitation energies,” J. Chem. Theory Comput. 19, 7983-7988 (2023).
M. R. Hennefarth, M. R. Hermes, D. G. Truhlar, and L. Gagliardi, “Analytic nuclear gradients for complete active space linearized pair-density functional theory,” J. Chem. Theory Comput. 20, 3637–3658 (2024)
M. R. Hennefarth, D. G. Truhlar, and L. Gagliardi, “Semiclassical Nonadiabatic Molecular Dynamics Using Linearized Pair-Density Functional Theory,” J. Chem. Theory Comput. 20, 8741-8748 (2024).
M. R. Hennefarth, Y. Kim, B. Jangid, J. Wardzala, M. R. Hermes, D. G. Truhlar, and L. Gagliardi, “MC-PDFT nuclear gradients and L-PDFT energies with meta and hybrid meta on-top functionals for ground- and excited-state geometry optimization and vertical excitation energies,” J. Chem. Theory Comput. 21, 7890-7902 (2025).